Visualisation, done honestly
bioflow doesn’t reinvent plotting. It runs the field-standard visual tools as stages and surfaces their own output — a report hub plus the tidy data, so you plot the rest.
Layer 1 · tidy data
One row per sample +
a results.json schema. Load it in R/Python and make any figure.
Layer 2 · overview
A metrics table + a circular genome map (GenoVi/Circos), the assembly graph (Bandage), QUAST/Icarus and fastp reports — embedded or linked, never redrawn.
Layer 3 · interactive
Standard BAM / BED / GFF outputs open straight in IGV — we delegate, we don’t lock you in.
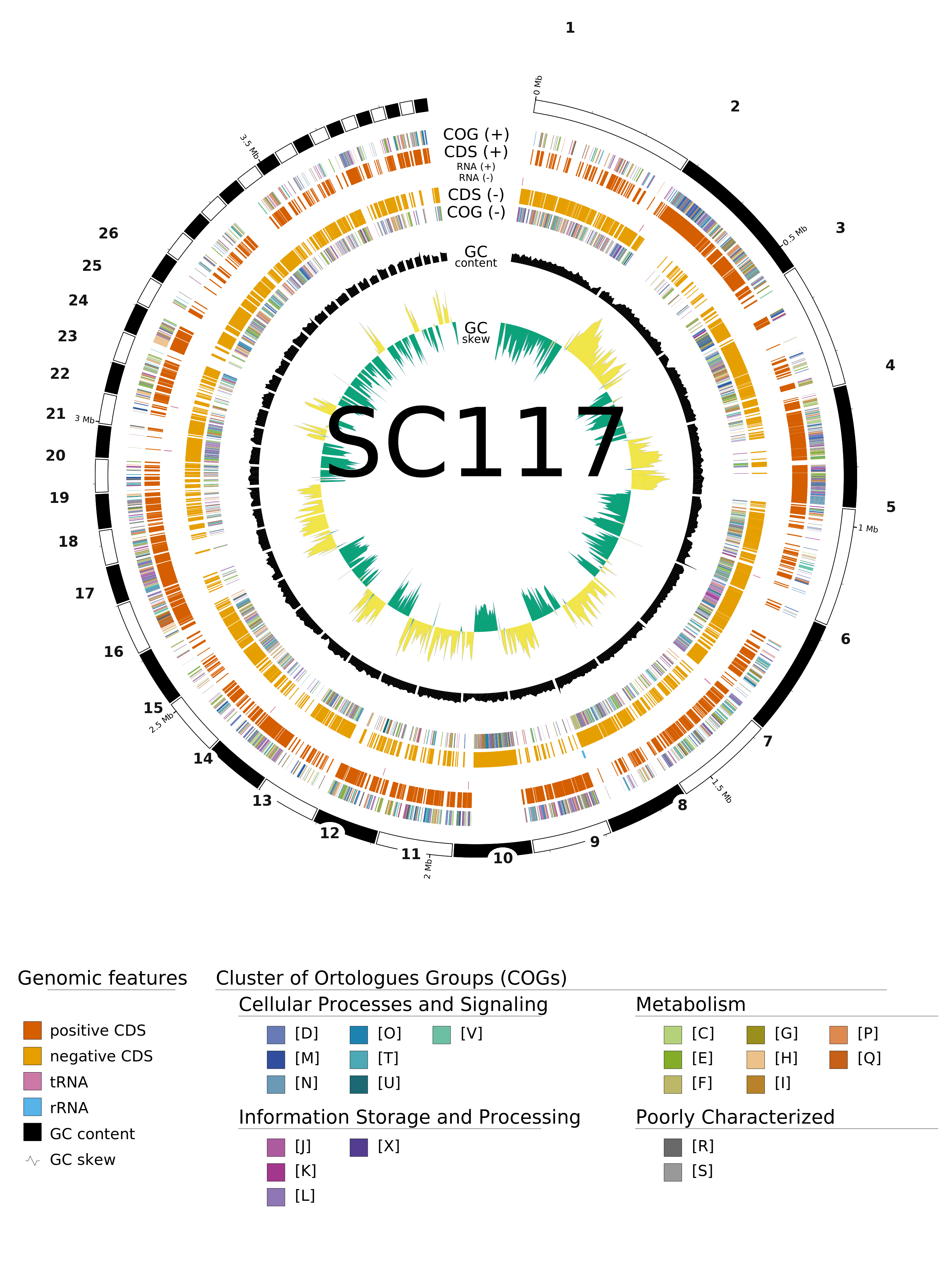
SC117 draft assembly. Rings, outermost to
innermost: contigs (numbered 1–26 by descending size, with a cumulative Mb scale);
protein-coding genes on the forward (+) and reverse (−) strands, coloured by COG functional
category; tRNA / rRNA; GC content (black, deviation from the genome mean); and GC skew
(green / yellow). Only contigs large enough to label are numbered; the full colour key is at
the foot of the figure. Rendered by GenoVi (Circos) from the Prokka annotation as the
pipeline's genome_plot stage — the actual, unedited output of a
bioflow recipe run.